Start date: 01-02-2022 End date: 31-07-2022
Clinical Problem
Deeper understanding of the immune system’s intricacies has led to clinical breakthroughs of personalized cancer vaccines in eliminating tumors in advanced-stage cancer patients. Formulated with patient’s tumor DNA fragments, cancer vaccines train a patient’s own immune system to recognize a patient’s mutated cancer proteins as ‘foreign’ and wage a lethal attack against tumors. The major puzzle in this field is: which of a patient’s hundreds of tumor mutations can trigger the immune system to attack tumors? Complementary to costly and time consuming wet-lab screenings (e.g., Sipuleucel-T was priced at $93,000), predictive algorithms that can quickly pinpoint neoantigens from a patient’s tumor-specific proteins are urgently needed, if personalized cancer vaccines are to be applied on a large scale. We aim to predict cancer vaccine candidates in this project. Our overall goal is to improve the efficacy, safety and development time of existing T cell based cancer vaccine approaches.
To serve as good cancer vaccine candidates, the tumor-specific peptides (i.e., short segments of tumor proteins) have to bind to the patient’s MHC (Major Histocompatibility Complex) molecule and form peptide:MHC protein complex in the endoplasmic reticulum. The complex is then shipped at the surface of the cell. If it is recognized by the T cell receptor (forming TCR-peptide:MHC complex), an immune response is triggered. T cells are constantly eliminating cancerous cells enforcing their physiological barrier role. Some cancerous cells presenting tumor-specific peptide:MHC complex are able to escape this defense mechanism and can lead to tumor growth. The effort is directed towards pinpointing peptides highly specific to tumors to facilitate cancer vaccine candidate discovery for therapeutic approach.
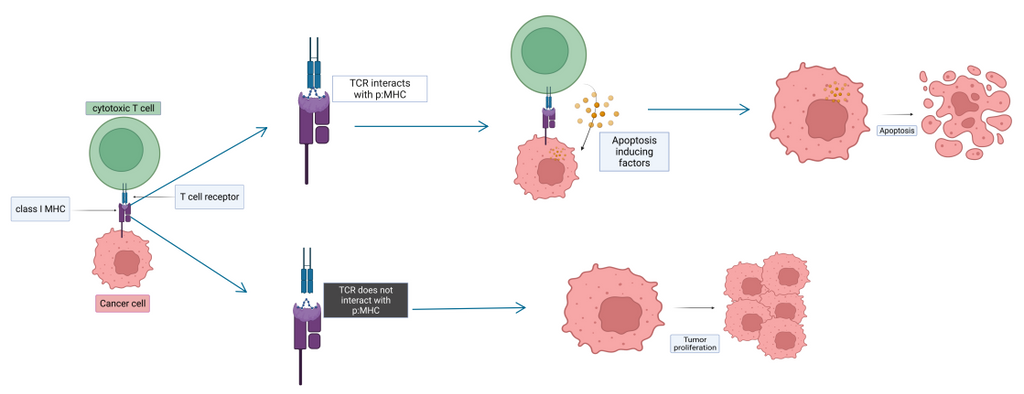
Solution
Today, state of the art predictive algorithms predict MHC-binding peptides based on the 1D sequences of MHC and peptides. They are used on a daily basis in laboratories to facilitate vaccine candidate discovery. Common residue patterns in the peptides’ sequence are elucidated by going over big datasets of experimental binding affinity values of peptide:MHC complexes, by using different computational approaches (e.g., fully connected neural networks, recurrent neural networks, hidden Markov models, etc). However, existing methods have limited performance. This may be caused by several factors. First, from a biological standpoint, the binding affinity depends on atomic features such as the hydrophobicity, Van der Waals interaction, solvent accessibility and other parameters. Sequence-based approaches can only indirectly capture the physico-chemical parameters that really determine the binding affinity of a similar complex. Second, the existing methods are data-driven ML methods. They rely heavily on the training data. This makes the existing methods to have limited accuracy on rare peptides that are not well-represented in the training data, for example,peptides coming from frameshift mutated proteins.
To tackle these problems, in this project we explored the 3D structure-based prediction approach and its generability to rare peptides. Our strategy is to train 3D-CNN on 3D peptide:MHC structures to learn interaction patterns in the space of energies, shapes and sequences. Our assumption is that energy and physico-chemical patterns dictating binding affinity learned by the trained model should be universally applicable and contain rich structural information. Specifically, the 3D CNN is trained on 3D models for peptide:MHC complexes generated with high-accuracy package (our PANDORA software). This will be done by using our 3D convolutional neural network framework, DeepRank. The solution provided allows to address the issue of predicting binding affinity between peptide and MHC with a novel structure-based approach, paving the way for further investigation concerning a structure based prediction on unseen peptide sequence data.
Data
The data used for this work comes from binding affinity assays. Several types of experiments are available to quantitatively assess how strongly one peptide can bind to the MHC groove. Experiments include measurements of half maximal inhibitory concentration (IC50), half maximal effective concentration (EC50), binding constant (Kd) which all allows us to assess the binding affinity. Some of these rely on the competitiveness of the peptide compared to a fluorescent/radioactive strong binder (EC50, IC50). Binding affinity assays are conducted in a completely artificial setup where one HLA allele is fixed on the support and the tested peptide is being added at varying concentration, in presence of a competitor. Resulting measurement is only viable if the conditions of the experiments are as close as possible to physiological chemical conditions (temperature, pH, electrolyte concentration). As a general rule, binding affinity values below 500nM indicates a binder (100nM and lower being strong binders), values above this threshold are not considered as binders.
As a proof of principle study, peptides with 9 residues (the most frequent length in nature) with known binding affinity to one MHC-I allele (HLA-A*02:01) are investigated. With these criteria, a total of 7,726 peptide:MHC-I (p:MHC-I) binding affinity values were retrieved. Among them, 4214 negatives (non-binders) and 3512 positives (binders), using 500nM as the cutoff.
Approach
For every binding affinity value, related p:MHC-I structure was generated using the p:MHC integrative modeling tool PANDORA. This tool takes as input the allele name of the MHC protein as well as the sequence of the peptide. PDB files containing the 3D structure of the peptide:MHC-I molecules are modeled by homology using templates from the PANDORA database. Next, using our DeepRank feature generator, first a 3D grid is built at the interface of the peptide:MHC-I encapsulating the interaction space. Typically, a grid of 30x30x30 Angstrom. Second, each atom-level and residue-level feature is calculated then mapped on the 3D grid. We train a 3D convolutional neural network on these 3D feature grids.
To compare with our structure-based method, we also trained a fully connected neural network (a multiple layer perceptron, MLP) on the same dataset using a BLOSUM62 encoded sequence of peptides. Here the data fed to the predictive model is a numerical representation of the peptide sequence concatenated with a sequence of 37 seven residues (pseudosequence) of the MHC known to be at the interface of the ppeptide:MHC complex. While the pseudosequence is of fixed length, the length of the peptide may vary from 8 to 15 residues. To insure same size representation, each peptide is represented by a concatenation of 3 paddings of length 15 resulting in a final representation of length 45: left-padded, center-padded and right padded. The BLOSUM62 encoding is a 21 by 82 matrix where each column is a vector of 21 numbers representing the BLOSUM62 permutation score for each amino acid. This way, each twenty amino acids (and empty X amino acid) are represented by a unique vector.
To evaluate the predictive performances of the 3D CNN as well as on the MLP on rare peptides, we clustered peptides based on their sequences, then trained and evaluated our predictors on these clusters of peptides. A set of clusters were used for training while one cluster was used for evaluation. This approach makes sure the peptides used for training have very distant sequences from the clusters used for evaluation of the model, thus simulating real life scenarios where tumor specific peptides are not found in the training data.
Results
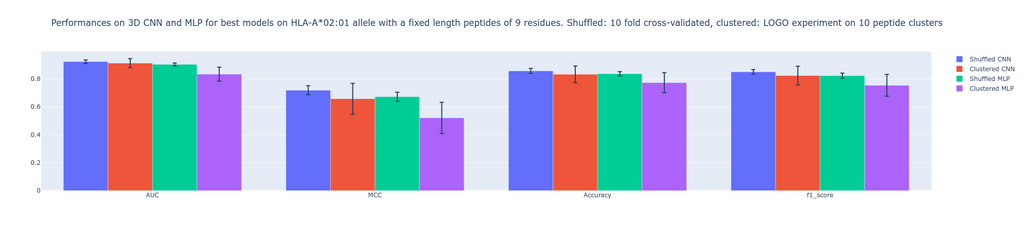
Metrics used to evaluate performances are 1)- Area under the receiver operating characteristic curve (AUC). AUC ranges from 0 to 1. Generally speaking, the higher the value the better the performance. For a value between 0-0.5, the false positive rate is higher than the true positive rate. The closer the value is to 1 the better the model is able to predict positive values while keeping the false positive rate low. The closer the value is to 0.5, the higher false positive rate gets. If the AUC is equal to 0.5, the amount of false positives is 50%. 2)- Mathew correlation coefficient MCC ranges between -1 and 1. Compared to AUC, it is a single-value classification metric incorporating sensitivity and specificity in the scoring process. A high score depends on a good sensitivity and specificity, together with false positive and negative rate being low. A score of 0 means random predictions, a score of 1 means perfect predictions and -1 opposite predictions. 3)- The F1 score gives insights into separation between precision and recall.
Our results show non significant visible differences when comparing these metrics. The MLP being inspired from an existing algorithm (MHCflurry 2.0) which uses BLOSUM62 encoded peptides is a good baseline for comparison. Indeed, the never used before structure-based prediction is able to perform as good as the MLP algorithm, which is a golden standard to predict binding affinity between peptide and MHC. This study first shows that 3D CNN applied to grids at the interface of peptide-MHC providing an extensive feature space is able to compete with MLP. Second, this study paves the way for further investigation concerning the performances of CNN compared to MLP given a bigger dataset. Note that we worked on a set of fixed-length peptides and on a single MHC allele. This is to the advantage of sequence-based approaches, which take fixed-length input. The advantage of our structure-based CNN is that it can naturally handle peptides with variable lengths, thus we expect this advantage to be further visible when we train and test on peptides with variable lengths and with diverse MHC alleles in a much bigger dataset.
The final report can be found here.



